语言

 直肠癌,铁死亡
直肠癌,铁死亡
 z-bio
z-bio
 2025-07-09
2025-07-09
 3656
3656
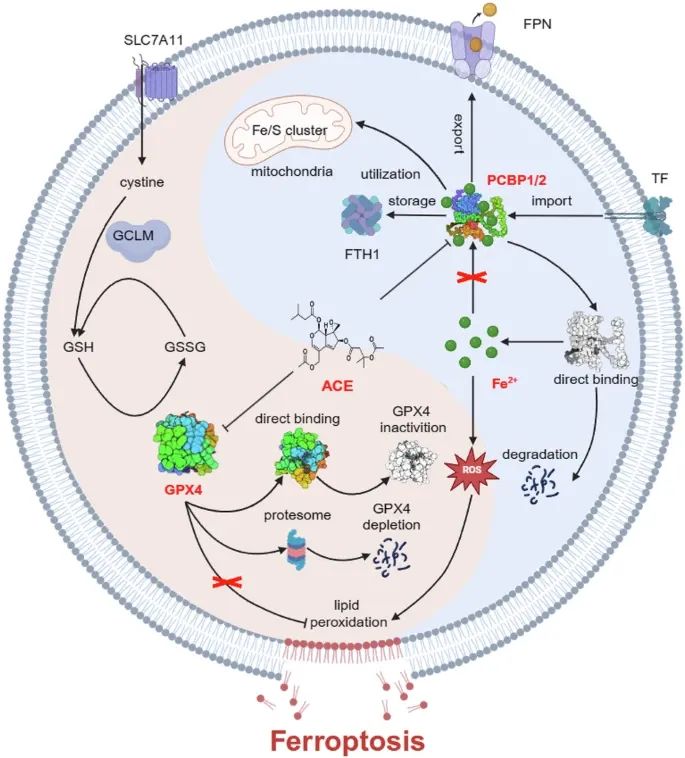
从一系列具有抗肿瘤活性的天然产物库中筛选出了缬草的acevaltrate(ACE)。在之前的研究中,ACE被证明对各种类型的癌症具有抗肿瘤作用。此外,乙酰戊酸盐已被证明通过HIF-1α, Otub1/c-Maf和USP10/CCND1轴诱导细胞凋亡和抑制增殖。因此,ACE被认为是一种有前景的天然抗肿瘤化合物。然而,其潜在的抗肿瘤机制尚不清楚。2025年7月7日,上海中医药大学交叉科学研究院张卫东/刘三宏教授团队在STTT(IF 52.7)发表题为“Acevaltrate targets PCBP1/2 and GPX4 to induce ferroptosis in colorectal cancer”的文章。天然产物乙酰缬草素acevaltrate (ACE),可以快速而强烈地诱导结直肠癌细胞的铁死亡。ACE不仅通过靶向铁伴侣蛋白PCBP1/2并降低其表达来增加结直肠癌细胞中的Fe2+水平,还通过靶向GPX4并抑制其酶活性来破坏结直肠癌细胞的抗氧化系统,导致其泛素介导的降解。这种双重作用使其诱导铁死亡的效果显著优于经典的铁死亡诱导剂。

摘要
由铁离子(Fe2+)和脂质过氧化蓄积诱导的铁死亡是一种新型的调节性细胞死亡形式,已成为肿瘤治疗研究的热点。识别可诱导肿瘤细胞铁死亡的小分子药物是一种非常有吸引力的治疗策略。在这里,我们筛选了一种天然产物乙酰缬草三酯acevaltrate (ACE),它可以快速而强烈地诱导结直肠癌细胞的铁死亡。ACE不仅通过靶向铁伴侣蛋白PCBP1/2并降低其表达来增加结直肠癌细胞中的Fe2+水平,还通过靶向GPX4并抑制其酶活性来破坏结直肠癌细胞的抗氧化系统,导致其泛素介导的降解。ACE的这种双重作用使其诱导铁死亡的效果显著优于经典的铁死亡诱导剂。我们的动物实验表明,ACE的治疗效果超过了已有的诱导铁死亡的药物,并且优于卡培他滨、TAS-102等一线临床药物。重要的是,ACE在结直肠肿瘤类器官中的抑制作用优于在细胞水平上的抑制作用,强调了其临床应用的潜力。本研究率先发现了同时靶向PCBP1/2和GPX4的小分子抑制剂,为通过铁死亡消除癌细胞提供了一种新的治疗策略。
1.ACE抑制结直肠癌细胞增殖,促进细胞死亡
受紫杉醇、奥沙利铂和5-FU的耐药性、转移和副作用问题以及用于癌症治疗的天然药物特异性和广谱优势的启发,我们选择RKO细胞,一种对细胞死亡敏感的低分化人结肠癌细胞系,在天然化合物库中筛选抗肿瘤活性。我们用不同的化合物(10 μM)孵育RKO细胞24 h,并通过细胞计数试剂盒(CCK-8)检测其活力。如图1a, b所示,使用抗肿瘤活性超过80%的标准,有13个化合物被鉴定为候选化合物。此外,通过测量不同肿瘤细胞系中ACE的半抑制浓度(IC50),我们确定ACE是具有广谱抗肿瘤作用的最佳化合物(图1c, d)。出乎意料的是,我们发现ACE对结直肠癌细胞更有效,IC50值范围为1.4 ~ 1.9 μM,对正常肠上皮细胞几乎无毒,提示ACE更适合用于结直肠癌的治疗(图1e-i)。
为了确定ACE的抗增殖作用,我们进行了集落形成实验和5-乙炔基-20-脱氧尿嘧啶核苷(EdU)实验,结果显示ACE显著降低了集落形成效率和细胞生长(图1j-m)。此外,流式细胞术显示ACE显著诱导HCT116和RKO细胞的G2/M期阻滞(图1n, o)。综上所述,ACE抑制细胞增殖,促进细胞周期阻滞,诱导细胞死亡,抑制细胞迁移和耐药。
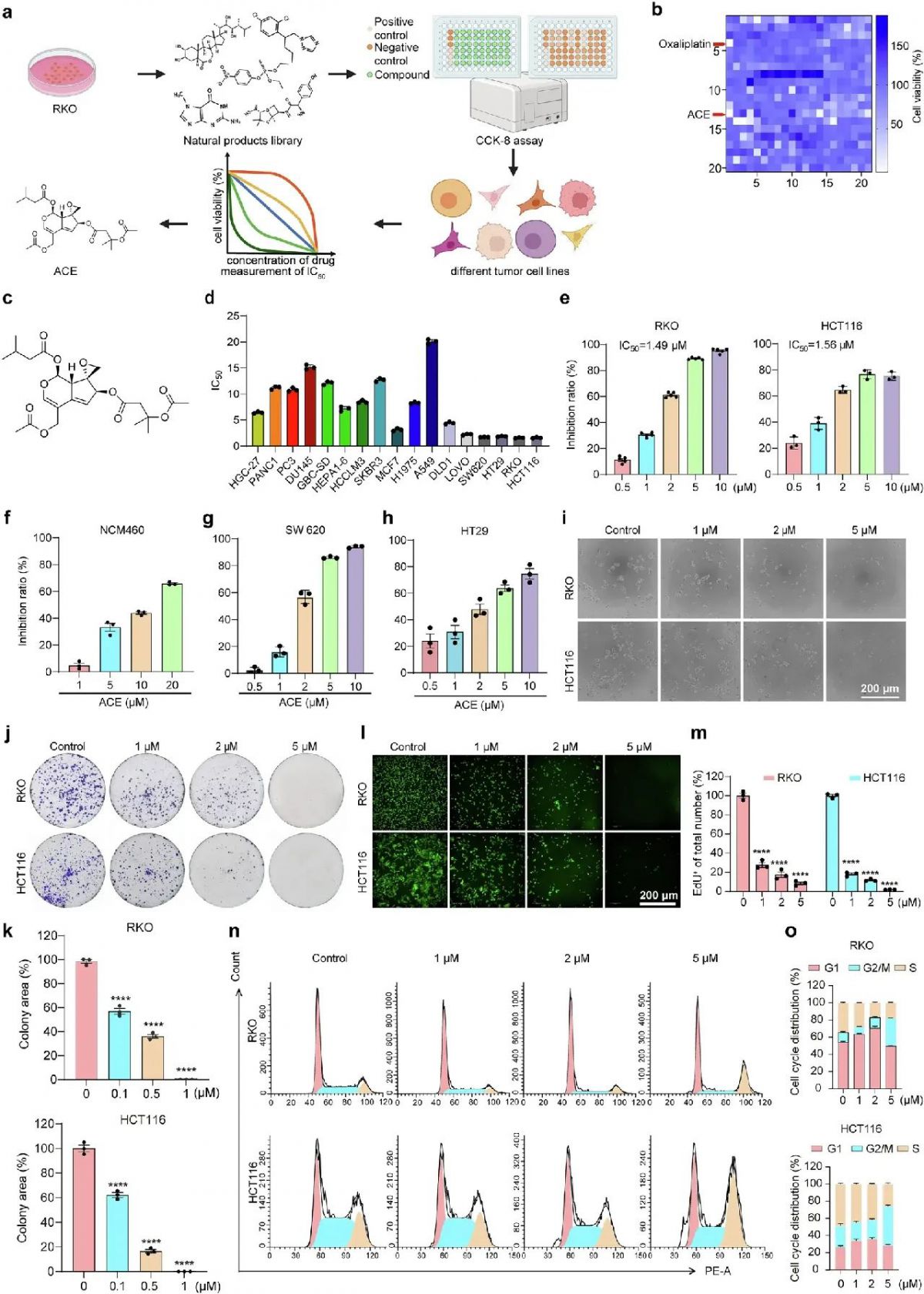
图1 ACE抑制细胞迁移和耐药
2.ACE诱导结直肠癌细胞发生特异性铁死亡
虽然ACE显著抑制细胞增殖并诱导细胞死亡,但与之前的报道不同,ACE并没有激活经典的凋亡途径。我们进一步探索了其潜在的非凋亡死亡机制。接下来,我们使用基于多组学的策略(蛋白质组学,转录组学和代谢组学)来研究ace诱导的细胞死亡的可能途径。对蛋白质组学数据进行KEGG富集分析发现,铁死亡通路是富集最多的通路,与铁死亡相关的泛醌生物合成和不饱和脂肪酸生物合成通路也在ACE处理组中富集(图2a、b)。正如预期的那样,转录组分析显示,ACE处理后,铁死亡信号通路被显著激活(图2c)。基于铁死亡在克服肿瘤耐药中的重要作用,我们初步假设ACE可能诱导结直肠癌细胞的铁死亡。
由于脂质过氧化对铁死亡至关重要,我们接下来分析了在ACE存在或不存在的情况下,RKO细胞中氧化脂肪酸和氧化还原剂水平的变化。首先,我们观察到ACE处理导致氧化多不饱和脂肪酸水平显著增加,尤其是花生四烯酸(AA)和亚油酸(LA),反映了细胞内脂质过氧化产物的积累(图2d, e)。此外,如图2f-i所示,ACE的细胞毒性可被铁死亡抑制剂阻断,而不被凋亡、坏死性凋亡或自噬抑制剂阻断。这些结果表明,ace诱导的特异性铁死亡是结直肠癌细胞死亡的关键决定因素,并且在很大程度上依赖于Fe2+。

图2 ACE可诱导结直肠癌细胞发生铁死亡
3.ACE诱导经典和非经典铁死亡
在显著富集铁死亡通路的基础上,我们通过形态学和生化特征来验证ACE是否诱导铁死亡。与其他形式的细胞死亡不同,脂质过氧化是铁死亡的一个关键标志物。用BODIPY-C11和Liperfluo两种荧光探针检测血管紧张素转换酶介导的脂质过氧化水平。BODIPY-C11染色显示,ACE促进脂质过氧化作用比铁死亡诱导性药物RSL3和erastin更强,这种作用被铁螯合剂DFO和脂质过氧化清除剂Lip-1逆转(图3a、b)。此外,ACE以浓度依赖性方式显著增加细胞内脂质过氧化水平(图3c)。此外,我们检测到细胞内MDA水平显著增加,MDA是脂质过氧化代谢产生的细胞毒性产物(图3d)。此外,透射电子显微镜显示线粒体变小,膜密度增加,嵴减少,这表明了铁死亡细胞的形态学特征(图3e)。此外,ACE剂量依赖性地破坏了线粒体功能,同时显著抑制了基础和最大细胞呼吸(图3f)。这些结果表明,ACE治疗显著增加了结直肠癌中脂质过氧化物的积累,进而诱导铁死亡。综上所述,这些数据表明ACE诱导经典铁死亡。
采用荧光探针(FerroOrange)和亚铁离子含量测定试剂盒检测细胞内游离的铁离子(Fe2+)。如图3g, h所示,ACE处理导致RKO和HCT116细胞中Fe2+的浓度显著增加,DFO显著逆转了ACE增加Fe2+水平的作用,而Lip-1没有。令人惊讶的是,与铁死亡诱导性因子RSL3和erastin相比,ACE显著增加了细胞内Fe2+水平(图3g)。此外,多组学结果显示,上调最显著的基因是HO-1,它与铁代谢相关。这一发现随后被Western blot实验证实(图3i)。当细胞内游离的亚铁离子在细胞质中蓄积时,通过Fenton反应产生ROS,导致肿瘤细胞铁死亡。如图3j所示,ACE处理后ROS水平显著升高并导致细胞死亡,而ROS抑制剂n -乙酰- l-半胱氨酸(NAC)和GSH逆转了ACE诱导的细胞死亡(图3k)。这些结果提示Fe2+在ACE诱导的铁死亡(非经典铁死亡)中起重要作用。综上所述,这些发现表明ACE通过经典和非经典途径诱导铁死亡。
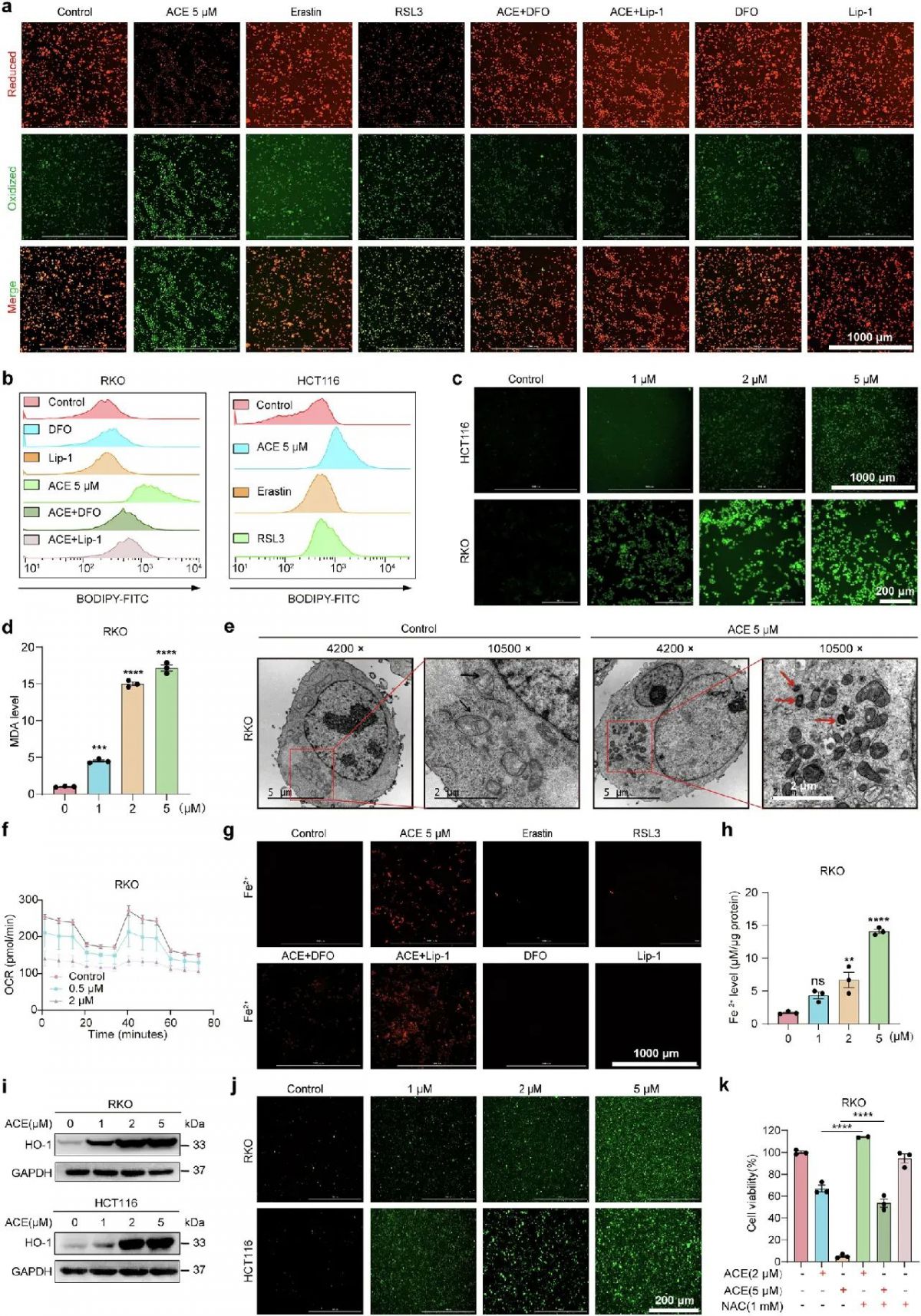
图3 ACE通过使GPX4失活并积累Fe2+诱导铁死亡
4.ACE抑制体内结直肠肿瘤生长
虽然ACE在结直肠癌细胞中显著抑制GPX4并升高Fe2+,从而诱导铁死亡,但这种化合物在体内的肿瘤细胞毒性尚不清楚。为了验证ACE的潜在体内抗肿瘤活性,我们对皮下接种HCT116-luc肿瘤的小鼠进行了玉米油或ACE口服治疗,每日1次,持续22天。25和50 mg/kg组的肿瘤荧光强度分别为对照组的57.5%和36.95%(图4a)。此外,与对照组相比,25和50 mg/kg的ACE治疗显著减小了肿瘤的大小、体积和重量(图4b-f)。各组间体重无明显差异,免疫组化结果也显示对心、肝、脾、肺、肾无明显毒性(图4d)。这些结果表明,ACE治疗显著抑制小鼠HCT116肿瘤的生长。随后,我们将RKO细胞接种到裸鼠体内,当肿瘤体积接近50 mm3时给予ACE。与其在细胞水平的效果相似,ACE在RKO荷瘤小鼠中的效果优于在HCT116小鼠中的效果(图4g-k)。
对肿瘤组织的免疫印迹分析显示,ace处理的肿瘤中ki67阳性细胞数明显低于对照组。此外,ACE没有激活caspase-3,表明ACE抑制小鼠肿瘤生长独立于凋亡(图4l)。为了确定ACE是否诱导肿瘤组织铁死亡,我们测定了脂质过氧化产物MDA和Fe2+的水平,发现ACE提高了MDA的水平,增加了Fe2+在小鼠肿瘤组织中的蓄积(图4m, n)。提示ACE可诱导肿瘤细胞发生铁死亡,有效抑制结直肠癌的发生。
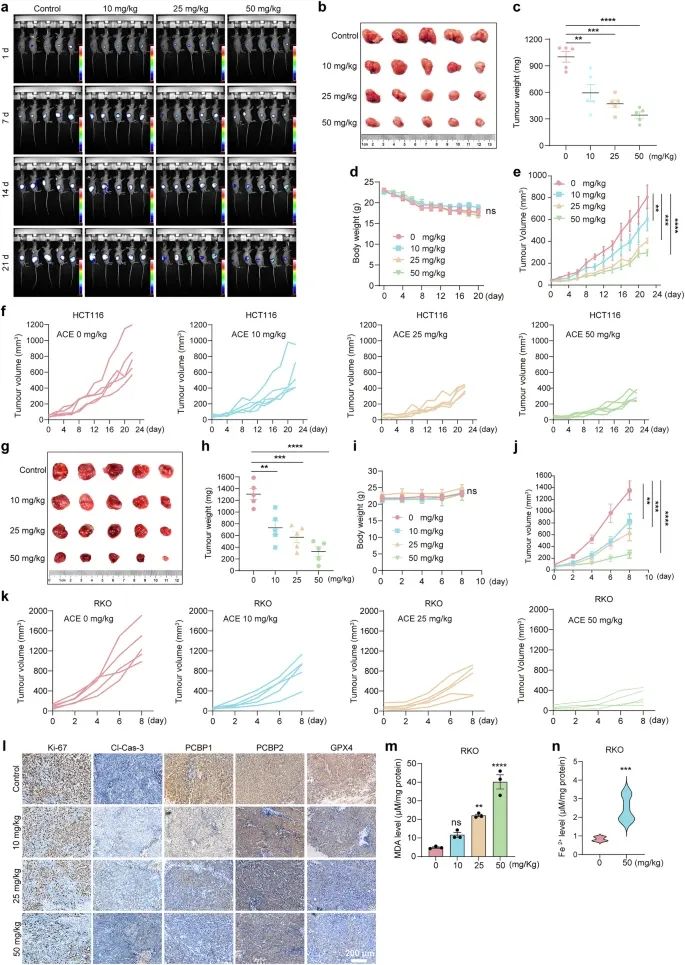
图4 ACE在体内诱导铁死亡
5.PCBP1/2的下调介导Fe2+释放并诱导铁死亡
为了探索ACE提高Fe2+水平的潜在机制,基于蛋白质组学结果,通过Western blot筛选与ACE相关的铁代谢蛋白(图5a, b)。此外,我们利用药物亲和力反应靶点稳定性(drug affinity responsive target stability, DARTS)技术检测小分子药物与靶蛋白的结合,探索ACE的潜在靶蛋白。DARTS预测了240个潜在靶蛋白(强度比≥1.2),其中4个基因(TF、GCLM、PCBP1和SLC39A14)与铁死亡直接相关(图5c)。值得注意的是,在铁代谢调节中具有相似功能的PCBP1或PCBP2在两种检测中均被富集。与多组学结果一致,PCBP1和PCBP2蛋白呈剂量和时间依赖性(1 h内)显著下调(图5a, b)。因此,我们推测ACE可能通过PCBP1和PCBP2增加Fe2+的释放。
为了验证ACE中升高的Fe2+水平与PCBP1/2之间的关系,我们进行了免疫荧光染色,发现PCBP1和PCBP2与细胞内Fe2+呈显著负相关(图5d)。接下来,我们设计了一个特异性小干扰RNA (siRNA)来模拟ACE对PCBP1/2的药理抑制作用。有趣的是,PCBP1和PCBP2敲低显著提高了Fe2+和ROS水平(图5e-g)。此外,在缺乏PCBP1/2的细胞中,ACE没有显著增加Fe2+水平(图5h, i)。此外,我们观察到敲低后的细胞状态非常差,流式细胞术结果表明结直肠癌细胞的细胞死亡增加(图5j)。这些结果表明,ACE可能通过PCBP1和PCBP2增加Fe2+和ROS来诱导铁死亡。如预期,PCBP1和PCBP2的过表达显著抑制了ACE增加脂质过氧化和Fe2+的能力,这进一步证实了我们的推测(图5k-m)。此外,在RKO细胞中过表达PCBP1和PCBP2可显著促进细胞增殖,显著抑制ACE的细胞毒性(图5n, o)。这些结果表明,ACE通过PCBP1和PCBP2诱导结直肠癌铁死亡。
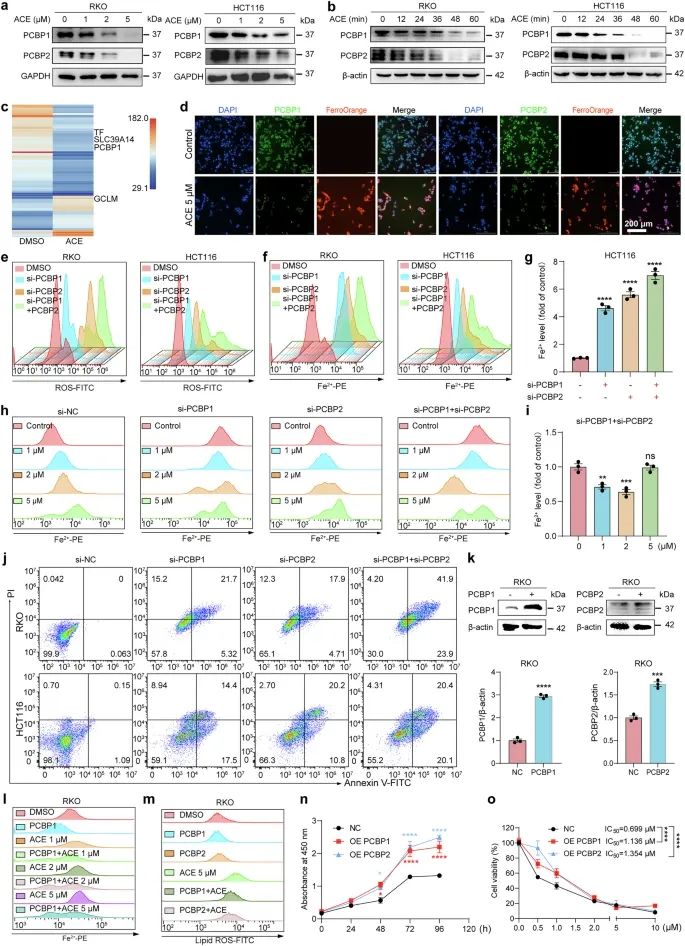
图5 PCBP1/2介导ace诱导的Fe2+蓄积和铁死亡
6.ACE与PCBP1/2结合诱导铁死亡
为了研究ACE是否直接与PCBP1和PCBP2结合,我们使用细胞热位移实验(CETSA)检测ACE处理后蛋白的热稳定性是否发生变化。如图6a, b所示,ACE处理后PCBP1和PCBP2蛋白的热稳定性降低,这与DARTS的结果一致。此外,我们通过表面等离子体共振(SPR)测定了PCBP1和ACE之间的亲和力,结果显示ACE直接与PCBP1结合,KD值为0.8464 μM(图6c)。此外,ACE从PCBP1蛋白中缓慢解离,表明ACE以更稳定的方式与PCBP蛋白结合(图6c)。此外,DTT,一个富含半胱氨酸的巯基供体,可以竞争与PCBP1/2的半胱氨酸依赖性ACE结合。如图6d, e所示,ACE引起的PCBP1/2的下调和细胞毒性的增加被过量DTT的加入部分逆转,表明ACE直接与PCBP的半胱氨酸残基结合。然后,我们使用Molecular Operating Environment(MOE)软件模拟ACE通过其半胱氨酸残基与PCBP1/2共价结合。通过对接模拟探索ACE与PCBP1 (AlphaFold ID AF-Q15365-F1)和PCBP2 (AlphaFold ID AF-Q15366-F1)各半胱氨酸残基位点的结合模式。如图6f所示,Cys54与ACE骨架之间形成了稳定的氢键,表明Cys54可能是PCBP1和PCBP2的结合位点。此外,Cys293可能是PCBP1的另一个结合位点。为了进一步证实ACE与PCBP1/2的直接结合,我们过表达了野生型PCBP1/2, Cys54突变型PCBP1, Cys293突变型PCBP1和带GFP标签的Cys54突变型PCBP2,随后通过微量热泳动(MST)分析了ACE与PCBP1/2的直接结合。正如预期的那样,ACE与PCBP1/2强结合,估计Kd值为4.64 nM和204 nM。当Cys293突变为丙氨酸时,其Kd是野生型PCBP1的325倍,而当Cys54突变为丙氨酸时,PCBP1和PCBP2均不能检测到ACE结合(图6h, i)。我们一致地发现,在过表达PCBP1/2 C54A的RKO细胞中,ACE的细胞毒性降低(图6j)。这些结果表明,ACE通过直接结合PCBP1和PCBP2,提高Fe2+水平,从而诱导结直肠癌铁死亡。
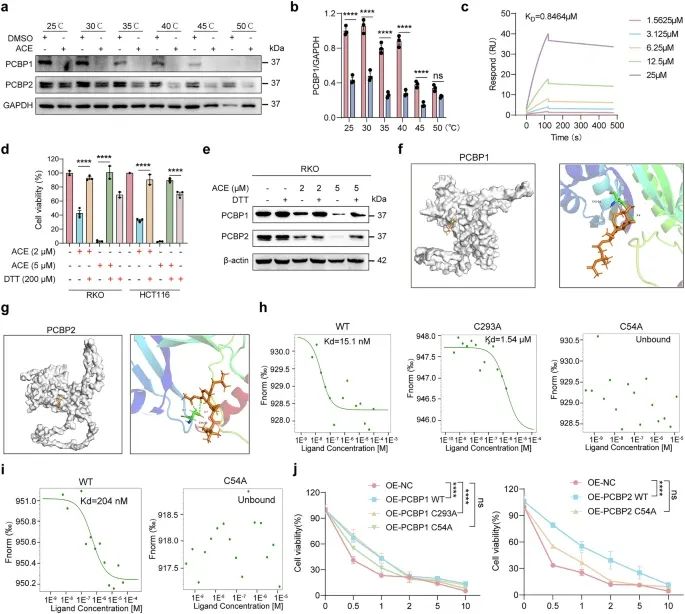
图6 ACE与PCBP1/2直接结合
7.ACE是一种天然的ⅱ类铁死亡诱导剂
由于在多组学数据中观察到一系列抗氧化基因和因子的上调,我们检测了一些抗氧化基因的表达变化。与多组学结果一致,ACE显著上调核因子红系衍生2样蛋白2 (Nrf2)及其下游靶基因(如SLC3A2、SLC7A11和GCLM)、铁死亡抑制蛋白1 (FSP1)和二氢乳清酸脱氢酶(DHODH)的表达。此外,我们发现铁死亡防御机制的核心蛋白GPX4的表达显著下调(图7a、b)。GPX4的失活可以通过两个过程发生:细胞内谷胱甘肽(GSH)的消耗或直接靶向GPX4。如图7c所示,ACE处理增加了RKO细胞中GSH和胱氨酸的水平。此外,ACE迅速增加ROS水平,抑制GPX酶活性,表明ACE可能像RSL3一样直接与GPX4结合(图7d)。正如预期的那样,缺乏GPX4显著促进了脂质过氧化积累和细胞死亡(图7e、f)。然而,GPX4过表达增加了RKO细胞中ACE的IC50值(图7g)。过量的DTT逆转了GPX4的下调,证实了ACE与GPX4的半胱氨酸残基结合(图7h)。如图7i所示,我们进行了类似的分子对接模拟,发现活性位点硒代半胱氨酸(U46)是ACE (Protein Data Bank ID 6NH3)与GPX4共价结合的最有利位点。此外,ACE和RSL3(阳性对照)处理后GPX4蛋白的热稳定性增加,表明ACE可以结合并稳定GPX4蛋白(图7j)。最后,我们在野生型和破坏性突变型U46中对gfp标记的GPX4进行了MST分析。ACE结合GPX4蛋白的Kd值估计为196 nM(图7i)。然而,一旦U46突变为丙氨酸,GPX4与ACE之间的结合消失,这进一步降低了ACE的细胞毒性(图7i)。这些结果表明,ACE通过直接结合GPX4中的U46而使GPX4失活,触发脂质过氧化和随后的铁死亡。
接下来,我们研究了ACE引发GPX4蛋白降解的机制。Western blot和real-time PCR结果显示,ACE不仅下调GPX4的表达,而且下调其mRNA水平(图7a, b)。在本研究中,我们主要研究GPX4的翻译后调节,因为GPX4的蛋白质水平比其RNA水平下降得更快(图7b)。如图7k, l所示,当与蛋白翻译抑制剂放线菌酮(cycloheximide, CHX)联合时,ACE处理的细胞中GPX4的半衰期比未处理的细胞更快,表明ACE主要在蛋白水平降解GPX4。此外,Western blot实验表明,蛋白酶体抑制剂MG132逆转了ACE对GPX4的降解,而CQ对GPX4的降解没有逆转作用(图7m)。此外,IP实验显示ACE增加GPX4的泛素化(图7n)。ACE通过泛素-蛋白酶体途径介导GPX4降解,诱导结直肠癌细胞发生铁死亡。
最后,RSL3的细胞毒性消失,而当GPX4被敲低时,ACE的细胞毒性部分降低,因为RSL3没有增加Fe2+(图7o, p)。如图4l所示,肿瘤组织中GPX4、PCBP1、PCBP2蛋白水平呈剂量依赖性降低,与体外实验结果一致。这些结果强调了ACE诱导结直肠癌细胞铁死亡的双重机制的有效性,即灭活GPX4和下调PCBP1/2释放Fe2+。
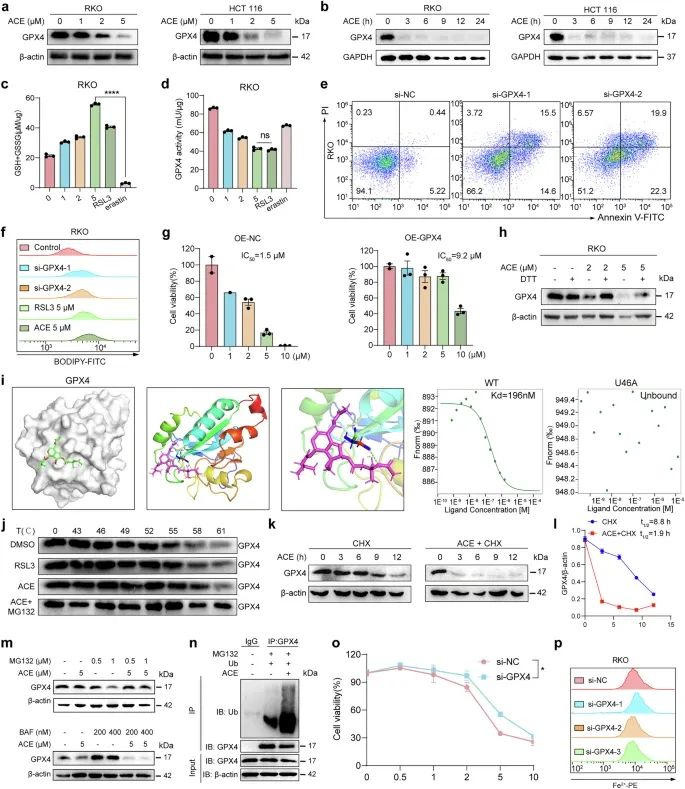
图7 ACE诱导GPX4耗竭导致铁死亡
8.PCBP1/2和GPX4在人肿瘤组织中的蛋白表达水平
为了验证上述模型用于临床肿瘤治疗的潜力,我们在异种移植模型中比较了ACE与临床一线药物和铁死亡阳性药物的肿瘤抑制效果。我们发现,ACE的肿瘤抑制作用显著大于铁死亡阳性药物索拉非尼和青蒿素,甚至优于临床一线药物卡培他滨和TAS-102,因为ACE升高Fe2+和降低PCBP2和GPX4的效果显著优于其他治疗(图8a-d)。虽然ACE通过靶向PCBP1/2和GPX4诱导肿瘤细胞发生铁死亡,但已有研究表明铁死亡可引起急性肝损伤和急性肾损伤。为了评估ACE治疗肿瘤的安全性,我们进一步进行了急性毒理学实验,在小鼠中口服ACE (50 mg/kg),每天10天。如补充表1所示,小鼠血液参数如红细胞、白细胞计数、血小板计数均无明显变化;肝肾损伤指标ALT、AST、BUN、Cr等均在正常范围内(图8e)。此外,我们检测了小鼠心脏、肝脏和肾脏组织中的Fe2+水平,发现ace处理组小鼠的心脏、肝脏和肾脏组织与对照组之间没有显著差异(图8f)。这些结果表明,治疗剂量的ACE在正常组织中没有明显的诱导铁死亡的风险,ACE的临床应用为转化医学提供了坚实的基础。
构建结直肠癌类器官模型,评估ACE的敏感性。用不同浓度的ACE(0.01、0.1、1、5、10、25、50 μM)培养6 d后,以绿色荧光为代表的活细胞大小明显减小,以红色荧光为代表的死细胞数量明显增多,表明类器官的生长受到明显抑制,如图8g所示。通过面积法测量类器官的灵敏度,ACE对3种类器官模型的IC50值分别为58、148和340 nM(图8h)。对结直肠癌患者样本的总生存期分析显示,高水平GPX4、PCBP1和PCBP2的患者总生存期显著较差(图8i)。此外,我们通过免疫荧光染色研究了PCBP1, PCBP2和GPX4在结肠癌患者中的蛋白表达(图8j)。这些发现提示PCBP1和PCBP2作为肿瘤诊断的生物标志物具有潜在的临床意义。
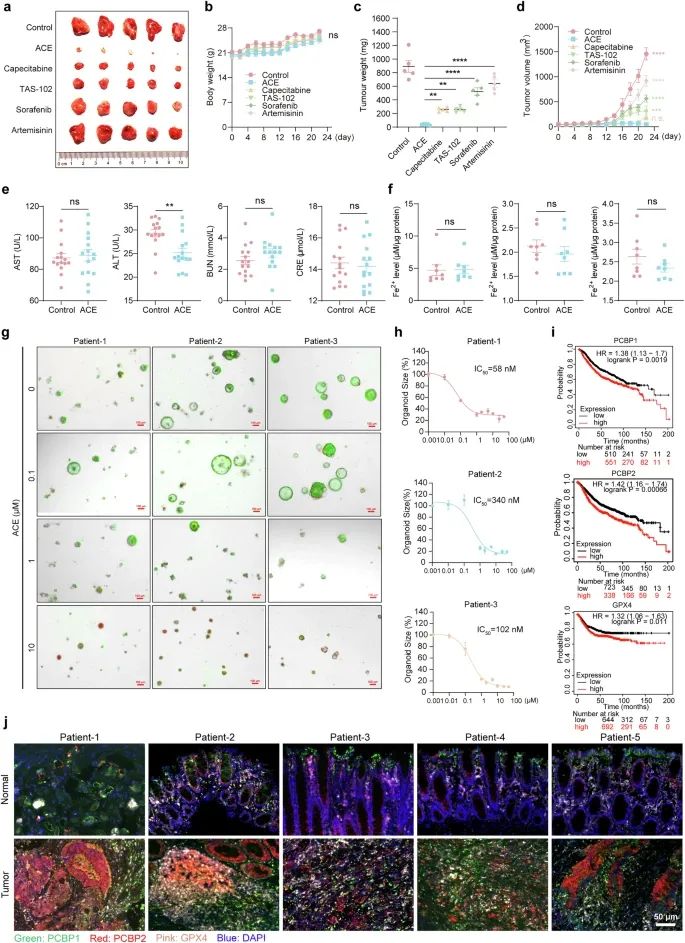
图8 靶向PCBP1/2作为潜在的肿瘤抑制策略
结论
综上所述,我们的研究表明ACE通过双重机制诱导肿瘤细胞的铁死亡:一方面,ACE通过直接结合和降解PCBP 1/2来增强细胞内Fe2+诱导的铁死亡,另一方面,ACE通过直接结合GPX4来诱导GPX4的泛素化和降解。此外,ACE在小鼠结肠癌模型甚至人类结肠癌类器官模型中均显示出较强的抗肿瘤作用。ACE的双靶点机制既规避了单靶点诱导剂的代偿性耐药问题,又通过天然产物的多靶点特性实现高效低毒的肿瘤选择性杀伤。我们期待这种双重机制为结直肠癌的临床管理提供新的策略。